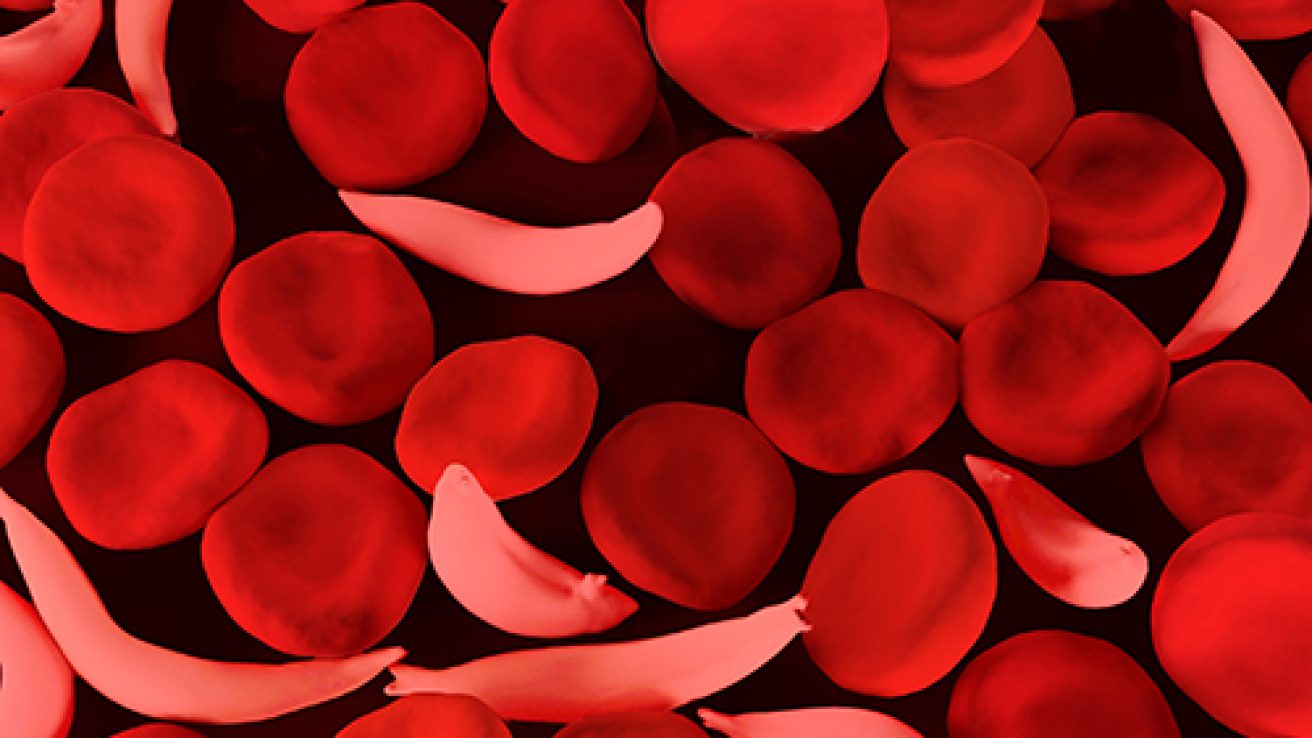
Sickle cell disease (SCD) is the most common genetic disease affecting 100,000 people in the U.S. According to the CDC 1 out of 365 African Americans and 1 out of 16,300 Hispanic- Americans have sickle cell disease. The majority of SCD patients are medicaid beneficiaries (less than 70% of doctors in the U.S. accept new medicaid beneficiaries). Below are current treatment options for sickle disease.
Voxelotor
The U.S. Food and Drug Administration (FDA) approved a new medicine in 2019 to treat sickle cell disease in adults and children 12 years and older. The oral medicine prevents red blood cells from forming the sickle shape and binding together. This may decrease the destruction of some red blood cells, which in turn lowers the risk for anemia and improves blood flow to your organs.
Possible side effects include headache, diarrhea, abdominal pain, nausea, fatigue, and fever. Rarely, allergic reactions may occur, causing rashes, hives, or mild shortness of breath. Talk to your doctor about other medicines you take.
Crizanlizumab-tmca
In 2019, the FDA also approved a new medicine to reduce the number of pain crises experienced by adults and children 16 years and older who have sickle cell disease. The medicine, which is given through an IV in the vein, helps prevent blood cells from sticking to blood vessel walls and causing blood flow blockage, inflammation, and pain crises.
Possible side effects include nausea, joint pain, back pain, and fever.
Penicillin
In children who have sickle cell disease, taking penicillin two times a day has been shown to reduce the chance of having a severe infection caused by the pneumococcus bacteria. Newborns need to take liquid penicillin. Older children can take tablets.
Many doctors will stop prescribing penicillin after a child has reached the age of 5. Some prefer to continue this antibiotic throughout life, particularly if a person has hemoglobin SS or hemoglobin Sβ0 thalassemia, since people who have sickle cell disease are still at risk. All people who have had surgical removal of the spleen, called a splenectomy, or a past infection with pneumococcus should keep taking penicillin throughout life.
Hydroxyurea
Hydroxyurea is an oral medicine that has been shown to reduce or prevent several sickle cell disease complications. This medicine was studied in patients who have sickle cell disease, because it was known to increase the amount of fetal hemoglobin (hemoglobin F) in the blood. Increased hemoglobin F provides some protection against the effects of hemoglobin S.
Hydroxyurea was later found to have several other benefits for people who have sickle cell disease, such as decreasing inflammation.
- Use in adults. Many studies of adults with hemoglobin SS or hemoglobin Sβ thalassemia showed that hydroxyurea reduced the number of episodes of pain crises and acute chest syndrome. It also improved anemia and decreased the need for transfusions and hospital admissions.
- Use in children. Studies in children with severe hemoglobin SS or Sβ thalassemia showed that hydroxyurea reduced the number of vaso-occlusive crises and hospitalizations. A study of children between the ages of 9 and 18 months with hemoglobin SS or Sβ thalassemia also showed that hydroxyurea reduced the number of pain episodes and dactylitis. There is no information about how safe or effective hydroxyurea is in children under 9 months of age.
- Pregnant women should not use hydroxyurea.
Since hydroxyurea can decrease several complications of sickle cell disease, most experts recommend that children and adults with hemoglobin SS or Sβ0 thalassemia who have frequent painful episodes, recurrent chest crises, or severe anemia take hydroxyurea daily.
Hydroxyurea can cause the blood’s white cell count or platelet count to drop. Rarely, it can worsen anemia. These side effects usually go away quickly if a patient stops taking the medicine. When the patient restarts it, the doctor usually prescribes a lower dose.
It is still unclear whether hydroxyurea can cause problems later in life in people who have sickle cell disease and take the medicine for many years. Studies so far suggest that it does not put people at a higher risk of cancer and does not affect growth in children, but further studies are needed.
Clinicians may recommend transfusion to treat and prevent certain sickle cell disease complications. These transfusions may include:
- Acute transfusion to treat complications that cause severe anemia. Doctors may also use transfusions when a patient has an acute stroke, in many cases of acute chest crises, and in multi-organ failure. A patient who has sickle cell disease usually receives blood transfusions before surgery, to prevent complications.
- Red blood cell transfusions to increase the number of red blood cells and provide normal red blood cells that are more flexible than red blood cells with sickle hemoglobin.
- Regular or ongoing blood transfusions for people who have had an acute stroke, to reduce the chances of having another stroke. Doctors also recommend blood transfusions for children who have abnormal transcranial Doppler (TCD) ultrasound results, because transfusions can reduce the chance of having a first stroke. Some doctors use this approach to treat complications that do not improve with hydroxyurea. Doctors may also use transfusions in people who have too many side effects from hydroxyurea. Possible complications include alloimmunization, which can make it hard to find a matching unit of blood for a future transfusion; infection; and iron overload.
A blood and bone marrow transplant is currently the only cure for sickle cell disease, but it is not for everyone. Most patients who have sickle cell disease either are too old for a transplant or do not have a relative who is a good enough genetic match to be a donor. A well-matched donor is needed for a patient to have the best chance for a successful transplant.
Most sickle cell disease transplants are currently performed in children who have had complications such as strokes, acute chest crises, and recurring pain crises. These transplants usually use a matched donor. Blood and bone marrow transplants are riskier in adults.
Several medical centers are researching new sickle cell disease blood and bone marrow transplant techniques in children and adults who do not have a matched donor in the family or who are older than most recipients. Hopefully, more people who have sickle cell disease will be able to receive a transplant in the future using these new methods.
Blood and bone marrow transplants are successful in about 85 percent of cases involving children when the donor is related and HLA (human leukocyte antigen)-matched. Even with this high success rate, transplants still have risks. Complications can include severe infections, seizures, and other clinical problems. About 5 percent of people who have received such transplants have died. Sometimes transplanted cells attack the recipient’s organs. This is called graft-versus-host disease. Medicines are given to prevent many of the complications, but they still can happen.
Source: